
 >
>短杆菌属海洋环境适应机制的泛基因组学
时间:
摘 要:【目的】为了探究短杆菌属对海洋环境的适应机制。【方法】本研究通过对 6 株分离自不同洋区、属于不同分类单元的短杆菌菌株进行测序、拼接和注释,结合 23 株从美国国家生物技术信息中心(NCBI)下载的短杆菌属模式菌株及非模式菌株的基因组数据,进行泛基因组学分析和物种进化分析。【结果】泛基因组学分析表明短杆菌属具有开放型泛基因组,这与该属生存环境多样性特征相符合。海洋来源的短杆菌与其他生境来源的短杆菌在基因组水平上表现出明显的差异性,主要是进化过程中的基因家族扩增收缩、转运蛋白家族、代谢通路和 CRISPR 等方面的差异。【结论】通过基因组水平上的差异初步揭示了海洋来源短杆菌对海洋环境的适应性,为深入了解短杆菌菌株的环境适应机制奠定了基础。
关键词:短杆菌属;泛基因组;比较基因组;海洋环境适应性
短杆菌属(Brevibacterium)属于放线菌门,是短杆菌科中有效种数目最多的一个属。1953 年 Breed 首次以 Brevibacterium linens 为模式种建立了该属[1]。目前该属有效发表的菌种有 35 个 (https://lpsn.dsmz.de/genus/brevibacterium) , 菌株分离自多种环境,例如大米[2]、奶酪[3]、人类皮肤[4]、受损的壁画[5],部分菌株是从发酵海鲜[6]、腐烂的褐藻体[7]和深海沉积物[8–9]等高盐度的海洋环境中分离获得。短杆菌属菌株在工业上具有很多应用,研究发现短杆菌属可以利用细胞外蛋白酶和脂肪酶分解脂类和蛋白质 (如酪蛋白) [10–11],同时,许多短杆菌属细菌也具有修饰含硫氨基酸以产生挥发性硫化物的能力,从而有利于挥发性风味物质的产生[12–14]。短杆菌属菌株常在许多不同类型的奶酪中用作表面成熟培养物,来缩短奶酪的成熟时间,减少变质,更好地控制奶酪香气并提高食品安全性[15]。除此之外,研究表明部分短杆菌还具有使合成染料脱色的功能[16]。在农业中,菌株 B. linens RS16 可通过调节抗氧化剂防御和 H+ ATPase 活性赋予水稻基因耐盐性[17]。 Brevibacterium antiquum DSM 21545T 被用于通过在混合培养的废水流中生长来回收富含磷的产品[18]。
由于短杆菌在工业奶酪制造、生物降解和污水处理等生物技术方面应用成果显著,越来越多的短杆菌完成了基因组测序,为了解短杆菌菌株的生物学特征提供了有力支撑。Tettelin 等[19]于 2005 年第一次提出了泛基因组(pan-genome)的概念。泛基因组分析能够获得同一物种内的许多个体的基因组,用以完善该物种的遗传信息,能够捕获只存在于单个或部分个体及种群基因组上的特异性序列和功能基因,为深入研究该物种的系统进化地位以及与形态差异相关的功能生物学奠定基础。本文以实验室前期分离鉴定的 6 株来自不同海域的短杆菌属菌株作为主要研究对象,结合从 NCBI 数据库下载的 23 株短杆菌属模式菌株或非模式菌株的基因组数据,采用泛基因组学方法,分析比较不同菌株之间的基因组特征。利用核心基因簇,研究菌株之间的演化关系。此外,结合基因组功能分析,探讨海洋短杆菌与其他生境中的短杆菌在基因组上的差异,探究短杆菌属对海洋环境的适应机制。
1 材料与方法
1.1 菌株来源及选择标准
本研究中所使用的短杆菌基因组数据主要是从 NCBI 下载,有模式菌株基因组数据的种优先下载该种的模式菌株基因组数据,没有模式菌株基因组数据的种,下载该种非模式菌株的相关基因组数据。目前,短杆菌属共有 35 个有效发表的种,在 NCBI 数据库中,已有 14 个种的模式菌株基因组数据被公开,另有 9 个种虽没有该种的模式菌株基因组数据,但该种中有非模式性菌株的相关基因组数据。因此,我们从 NCBI 下载了这 14 个种的模式菌株及另外 9 个种中的 9 个非模式菌株的基因组数据,共计 23 个种的相关菌株基因组数据进行后续研究。此外,本实验室也从不同洋区获得了大量的短杆菌属菌株,以 16S rRNA 基因序列相似性 99%为标准共划分为 6 个操作分类单元 (operational taxonomic units,OTU),从每个 OTU 中各选取了 1 株菌株(YB235、Wo024、 WW007、O1、o2、W7.2)作为代表性菌株进行了全基因测序,其中菌株 O1 已完成新种鉴定并公布了有效名,YB235、Wo024、W7.2 和 o2 为潜在的新种。29 株短杆菌的具体相关信息见表 1,其中有 9 株菌来源于海洋,称为海洋来源菌株,其他 20 个菌株来自多个生境(人、土壤、奶酪和肉汤等),称为多重来源菌株。
1.2 基因组测序、组装和注释
本实验所分离的 6 株海洋来源的短杆菌完整基因组由天津生物芯片公司采用第三代测序技术 PacBio Sequel 系统测序完成。利用 HGAP 4[20]软件进行 De novo 组装获得。为了避免由于各项目所采用的基因预测及注释方法不同而造成的偏差,我们对 29 株菌采用 Prokka1.13 进行了重预测。同样,利用 BLAST 分别对 nr 库和 COG 直系同源蛋白数据库进行基因注释和 COG 注释,利用软件 Canoco5 对基因组大小、CDS 和 GC 含量进行主成分分析。利用 CRISPRCasFinder[21],在线预测各个菌株中的 CRISPR 元件,统计每个菌株中 CRISPR 元件的数量。
1.3 泛基因组学分析
1.3.1 泛基因组特征分析利用原核生物泛基因组自动化分析软件 (pan-genomics analysis pipline , PGAP)[22] 对 29 株短杆菌进行泛基因组分析。首先,根据流程中的格式要求,编写 python 程序,依据 Prokka 的注释结果,准备核酸(nuc)、蛋白(pep)和功能 (function)文件。功能聚类选择 GF 方法进行直系同源基因鉴定,将聚类结果 Orthologs_ Cluster.txt 输入 PanGP 绘制泛基因组特征曲线和核心基因组特征曲线[23]。我们从 PGAP 功能基因的聚类结果中,统计了各个菌株的核心基因、非必需基因以及特异性基因的分布等信息。
1.3.2 核心基因组建树、分化时间鉴定及 ANI (average nucleotide identity)分析
为更深入了解 29 株短杆菌的进化关系,我们挑选与短杆菌属同科不同属的 Spelaeicoccus albus D3-40T (accession:JACBZP000000000)菌株作为外源。对这 30 个菌株进行 PGAP 分析,得到单拷贝核心基因;利用 MAFFT v7.471 进行多序列比对[24],并用 TrimAL 切割多序列比对结果;利用软件 IQ-TREE1.6.12 筛选碱基替换模型并构建系统进化树[25]。
从 TIMETREE (http://www.timetree.org/)上获取不同物种的分化时间,然后利用 r8s 软件在系统进化树的基础上对菌株系统发育的分化时间进行鉴定[26]。
利用 perl 脚本 ANI.pl (https://github.com/ jhbadger/scripts/blob/master/ANI.pl)计算 29 个短杆菌相互之间的 ANI,并通过 Heml 软件绘制热图。
1.3.3 单拷贝核心基因一致性分析
将 29 株短杆菌分成海洋来源和多重来源两大分支,比较 2 个分支内部以及 2 个分支之间短杆菌基因序列的一致性,利用 blastn 对物种中单拷贝核心基因序列进行 all to all 比对。分别提取出两个分支内序列一致性大于 70%的比对结果,用 python 绘制序列一致性的密度分布曲线来观察两个分支内部基因序列的一致性差异。
1.3.4 转运系统
为研究不同环境下短杆菌与环境的相互作用,我们比较了海洋来源和多重来源两个分支短杆菌中转运蛋白系统的差异。提取注释为转运蛋白 (transporter protein) 和假定蛋白 (hypothetical protein)的基因作为转运蛋白的候选集,编写 python 脚本,下载 TransportDB 数据库中所有的 fasta 序列,通过 BLAST 比对 (identity 大于 50%,evalue 值 1e-5,如果一个基因比对上多个功能基因,选取 identity 最大的一个),进一步注释各个候选集中的转运蛋白。然后根据 TransportDB 中的转运蛋白家族功能分类对各个菌株中注释后的转运蛋白进行分类并统计数量,绘制热图比较分析具有差异的转运蛋白家族。
1.3.5 KEGG 代谢通路分析
为研究短杆菌属在不同环境下的信号通路,利用 KEGG 注释网站(https://www.genome. jp/kegg/kaas/)对各个菌株进行了在线的 KEGG 代谢通路分析,然后编写 python 脚本下载菌株所有通路的结果。比较细菌的信号系统、双组分系统以及磷酸转移酶系统 3 个与环境适应相关的通路[27]。
1.3.6 基因家族扩增和收缩
为研究短杆菌属在不同环境下的基因家族的变化,利用 Orthofinder2.3.3 分析短杆菌属所有菌株的基因组[28],获取统计基因家族中基因数量的文件。据此,用 CAFÉ 软件计算分析短杆菌属基因家族的扩增和收缩[29]。
2 结果与分析
2.1 基因组组装和注释
对 29 株短杆菌的注释结果如表 1,本实验室所测 6 株菌株均组装成 1 条完整的 contig,说明组装质量比较好,基因组大小在 4 Mb 左右, GC 含量等于或大于 0.65,编码 DNA 序列(coding DNA sequences,CDS)的数量为 2 020–4 041。
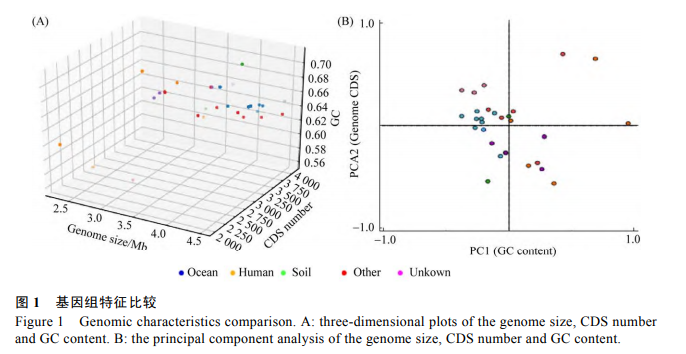
2.2 基因组特征比较
为研究不同环境下短杆菌基因组基本特征的差异,我们对 29株短杆菌的基因组大小、CDS 数量和 GC 含量进行比较(图 1A),发现海洋来源的短杆菌普遍具有较大的基因组,对以上三者进行主成分分析(图 1B),发现 CDS 和基因组大小可以比较明显的将海洋短杆菌和部分其他生境来源的短杆菌分开。
规律间隔成簇短回文重复序列(clustered regularly interspaced short palindromic repeat, CRISPR)是一种特异性的防御机制,能够有效防御质粒或噬菌体对细菌本身的水平基因转移,保持自身遗传信息的完整性。短杆菌属 CRISPR 分析发现,所有短杆菌均含有 CRISPR 元件。海洋来源短杆菌和多重来源短杆菌含有的 CRISPR 数量的均值都为 4。在 9 株海洋来源短杆菌株中,有 2/3 的菌株含有的 CRISPR 元件数量大于等于平均值;而 20 株其他生境来源的短杆菌有 1/2 菌株 CRISPR 元件数量大于等于平均值,说明大部分海洋来源短杆菌含有更多的 CRISPR 元件。这可能是因为海洋环境中含有更多的噬菌体,因此海洋来源的短杆菌需要进化出更多的 CRISPR 元件,以抵御外源 DNA 的侵害,降低水平基因转移。
2.3 泛基因学分析
2.3.1 泛基因组大小及其特征曲线
为了直观展示 29 株短杆菌泛基因组特征,根据 PGAP 聚类结果,利用 PanGP 绘制短杆菌的泛基因组特征曲线(图 2),蓝色表示泛基因组特征曲线,绿色则表示核心基因组特征曲线。可以看出,随着菌株数量增多,泛基因组呈现明显增大趋势,因此短杆菌具有开放型泛基因组,同时随着短杆菌数量增加核心基因组明显减小。这 29 株短杆菌的泛基因组大小为 17 445。泛基因组的特征方程为 y=2 561.89x 0.55+696.79,同时 R2 =0.999 946、平均每增加一个基因组就增加 500 多个基因。通常认为细菌需要改变自身的基因组,适应不同的环境,因此在多种不同的生境中生存的细菌需要一个更大的泛基因组,由于短杆菌生境的多样性,短杆菌属开放型的泛基因组与其生境多样性一致。
对海洋来源分支和多重来源分支分别进行泛基因组分析,可以看出两分支内也具有开放型泛基因组(图 2B、C),泛基因组的特征方程分别为 y=1 823.19x 0.53+1 762.53 且 R2 =0.999 901 和 y=3 034.61x 0.53+43.88 且 R2 =0.999 957。
2.3.2 核心基因及菌株特异性基因组分布
为研究短杆菌属的基因组的差异,我们分析了各个菌株中核心基因和菌株特异基因的分布。PAGP 功能聚类结果显示,29 株菌株共有的核心基因为 838 个,其中 734 个是单拷贝核心基因(图 3A)。分析两个分支内部核心基因和特有基因,海洋分支来源短杆菌共有的功能基因有 2 042 个,其中 1 912 个是单拷贝核心基因 (图 3B)。多重分支来源短杆菌共有的功能基因有 857个,其中 757个是单拷贝核心基因(图 3C)。将 29 株短杆菌特异性基因的分布进行比较,两个分支内各个菌株中特异性基因的数量较多,这是由于随着菌株数量的增加,一部分菌株特异性基因变成非必需基因。
2.3.3 进化分析
为了深入研究短杆菌属进化关系,以菌株 Spelaeicoccus albus D3-40T 作为外群,以 30 个物种共有的 640 个单拷贝核心基因构建系统进化树。借助软件 IQ-TREE 测试了 748 个碱基替换模型,最优模型是 Blosum62+F+R4,之后利用最大似然法,进行 1 000 次自展,成功构建了核心基因组系统进化树(图 4A)。同时构建了 30 个菌株的 16S rRNA 基因序列进化树(图 4B)。二者的拓扑结构并不一致,在核心基因组树中,基本所有分支的 bootstrap 值均为 100,而 16S rRNA 基因树只有 3 个,说明核心基因组树具有更大的可靠性。
在核心基因组树的基础上,探索短杆菌属的分化时间,发现短杆菌属最近共同祖先最早出现在大约 88 百万年前(图 5)。菌株 B. siliguriense DSM 23676T 和 Brevibacterium sp. o2 分化时间最近,在 100 万年前。
平均核苷酸相似度 (average nucleotide identity,ANI)是在核苷酸水平上比较两个基因组亲缘关系的指标,在近源物种之间具有较高的相似性,我们将 29 个短杆菌进行相互比较,计算 ANI。结果如图 6 所示,可以看出 B. iodinum、 B. sediminis、B. permense 的 ANI 聚类在一起,说明它们之间的遗传距离比较接近。
2.3.4 单拷贝核心基因序列一致性
为了研究不同生境下短杆菌基因序列的差异,我们提取短杆菌核心基因,分成海洋和多重来源两大分支。在两大分支内部,不同物种的核心基因序列进行相互比较,统计序列一致性。结果如图 7 所示,海洋来源的短杆菌核心基因一致性密度分布曲线的峰值约为 87%,而多重来源分支一致性密度曲线峰值约为 83%。海洋来源分支核心基因更保守,而多重来源分支核心基因具有更高的多样性以适应不同的生存环境。
2.3.5 转运蛋白家族比较
为了研究短杆菌和环境之间的相互作用,我们分析比较了海洋来源和多重来源跨膜运输系统的差异。我们根据 TransportDB 数据库中转运蛋白家族的分类,对两个分支富集的转运蛋白进行了比较(图 8)。海洋来源均含有的转运蛋白有 ABC (The ATP-binding cassette superfamily) 、 BCCT (The betaine/carnitine/ choline transporter (BCCT) Family)、MFS (The major facilitator superfamily)、SSS (The solute: sodium symporter family)、Nramp (The metal ion transporter family)、MgtE (The Mg2+ transporter-E family) 、 CitMHS (The citrate-Mg2+ : H+ citrate-Ca2+ :H+ symporter family)、NSS (The neurotransmitter:sodium symporter family)、 NCS2 (The nucleobase : cation symporter-2 family)、PiT (The inorganic phosphate transporter family)、BASS (The bile acid:Na+ symporter family),基本含有的是 Sulp (The sulfate permease family)、TRAP-T (The tripartite ATP-independent periplasmic transporter family) 、 DASS (The divalent anion:Na+ symporter family)。海洋来源富集更多 ABC、MFS、BCCT、SSS、Nramp、 MgtE、CitMHS、DASS 等转运蛋白。其中 BCCT 有助于协助相容性溶质甜菜碱,肉毒碱和胆碱的积累。这不仅有利于维持细胞渗透平衡,而且还充当蛋白质和细胞成分的稳定剂,防止变性,抵制高离子强度的影响。海洋来源菌株含有更多与营养吸收相关的转运蛋白:SSS、 NCS2。ABC 转运蛋白可能与其需要转运和代谢各种有毒物质和几丁质、支链淀粉、纤维素和淀粉能力相关。MFS 促进糖、药物分子、肽、三羧酸循环代谢产物、有机阴离子和无机阴离子等溶质在电化学梯度下进行跨膜运输[30]。
2.3.6 KEGG 代谢通路
为研究短杆菌如何针对不同环境做出适应性反应,针对各个短杆菌的 KEGG 代谢信号通路进行了分析。主要比较了两个分支中的双组分系统(two-component system)、细菌分泌系统 (bacterial secretion system)以及磷酸转移酶系统 (phosphotransferase system)。结果显示,在细菌分泌系统中,所有短杆菌均含有 SecDF、SecY、 YidC、SecA、FstY、ffh、TatC。部分海洋来源短杆菌(O1、o2、Wo024、YB235、FXJ8.269) 含有 SecE 基因,只有少部分多重来源短杆菌含有 SecE 基因(NBRC 14811、NCTC9742、ATCC 19391s、VKM Ac-2280、DSM 23676)。Sec 途径是细菌中第一个发现的跨细胞质膜向细菌周质和外膜输出蛋白质的分泌途径[31]。SecE 基因是一个与细胞胞外运输相关的基因[32],这可能是因为大部分海洋来源短杆菌向周围环境释放一类大分子的蛋白杂多糖胞外聚合物,防止冰晶形成,以适应低温环境。此外,在双组分系统中,只有 O1 同时含有与盐压力相关的 DegS 基因,与低温相关的 DesK 基因,细胞壁压力刺激物 VraS 基因,细胞壁活性物 LiaS 基因。这可能是因为短杆菌 O1 生活在 7 000 m 下的海底,这些基因可以帮助其适应高压、低温的环境。磷酸烯醇式丙酮酸依赖的糖磷酸转移酶系统(PTS)是一个大的碳水化合物活性转运系统,可以催化底物葡萄糖苷和葡萄糖、海藻糖磷酸化。我们发现含有 SecE 基因的菌株 (O1、o2、YB235、DSM 21545、FXJ8.269、 FDAARGOS_902、NBRC 14811、VKM Ac-2280、 DSM 22082、DSM 23676)基本含有与葡萄糖苷和葡萄糖、海藻糖磷酸化相关基因,可能是这些菌株为适应生存而在进化过程中获得的一种自身调节机制,可以更高效地利用碳源。
2.3.7 基因家族扩增和收缩
为研究短杆菌在进化过程中基因家族的变化,我们做了基因家族的扩张和收缩分析 (图 9)。最近共同祖先含有的基因家族有 5 966 个,在短杆菌进化过程中大部分基因家族都在收缩,在 55 百万年、50 百万年、41 百万年、 35 百万年、30 百万年分支节点的时候,基因家族收缩得最剧烈,从而导致大量的基因丢失,而海洋来源的短杆菌基本不在这些分支上(图 5,图 9),且大部分海洋来源短杆菌近期也没有经历大规模基因家族的收缩,这可能就是海洋来源短杆菌基因组偏大的原因。
3 总结
通过对短杆菌属不同生境菌株基因组进行比较分析,短杆菌基因组大小变化范围是 2.4–4.9 Mb。与多重来源短杆菌基因组相比,海洋来源短杆菌基因组偏大。这可能与多重来源短杆菌经历更多的基因家族收缩相关。比较两个分支的核心基因的一致性密度曲线,发现海洋来源短杆菌具有更高的一致性,而多重来源短杆菌具有更高的多样性,这可能是短杆菌属菌株为适应不同的生存环境,其核心基因的大小随环境的变化而调整导致的。
在转运蛋白家族上,海洋来源短杆菌富集更多的 ABC、MFS、BCCT、SSS、Nramp、MgtE、 CitMHS、DASS 及 BCCT 转运蛋白家族成员,这些物质有助于协助相容性溶质甜菜碱、肉碱和胆碱的积累。这些兼容的溶质可维持细胞渗透平衡,充当蛋白质和细胞成分的稳定剂。分析 KEGG 代谢路径,发现部分短杆菌菌株为适应海洋环境进化出了自己独特的基因以适应高压、寒冷的环境,同时为了更加高效地利用碳源获得了与 PTS 代谢相关的基因。海洋来源短杆菌也具有更多的 CRISPR 元件以应对海洋中大量噬菌体的威胁。
综上,本研究通过比较海洋来源与其他生境来源的短杆菌在基因组水平上的差异性,主要是进化过程中的基因家族扩增收缩、转运蛋白家族、代谢通路和 CRISPR 等方面的差异揭示了短杆菌的海洋环境适应性机制。下一步可对特定基因进行相关实验验证,深入分析短杆菌的海洋环境适应性机制。——论文作者:裴盛祥,黄小云,牛四文,谢富全,张改云
