
 >
>蛋白激酶Pbk在缺氧神经胶质细胞中的变化及与IGF-1的相关性
时间:
【摘要】目的探讨神经胶质细胞缺氧缺糖后不同时间点蛋白激酶Pbk的变化及与胰岛素样生长因子-1(insulin-likegrowthfactors-1,IGF-1)的相关性。方法培养BV2小胶质细胞、分离原代星形胶质细胞,制备氧糖剥夺(oxygenandglucosedeprivation,OGD)模型,采用反转录聚合酶链反应(reversetranscription-polymerasechainreaction,RT-PCR)技术观察信号分子Pbk、组蛋白去乙酰化酶2(histonedeacetylases2,HDAC2)以及IGF-1在两种胶质细胞OGD处理1、3、6h后的表达情况,并对信号分子和细胞因子进行了Pearson相关性分析。结果随着OGD时间延长,Pbk的转录水平在小胶质细胞中先升后降,在星形胶质细胞中逐渐升高,组间差异有统计学意义(P<0.05)。HDAC2的转录水平在小胶质细胞中逐渐下降,在星形胶质细胞中逐渐升高,组间差异有统计学意义(P<0.05)。IGF-1的转录水平在小胶质细胞中逐渐下降,在星形胶质细胞中逐渐升高,组间差异有统计学意义(P<0.05)。细胞缺氧缺糖后信号分子Pbk以及HDAC2在小胶质细胞和星形胶质细胞中的转录水平与IGF-1的转录水平均呈正相关关系(P<0.05)。结论缺血缺氧性损伤早期营养因子IGF-1在小胶质细胞中的分泌减少,在星形胶质细胞中的分泌增加,可能与Pbk/HDAC2/IGF-1这一通路激活相关。
【关键词】缺血缺氧;小胶质细胞;星形胶质细胞;细胞因子;PDZ连接激酶
神经胶质细胞是神经组织中除神经元以外的另一大类细胞,主要有星形胶质细胞和小胶质细胞等,可以分泌多种神经活性物质。胰岛素样生长因子-1(insulin-likegrowthfactors,IGF-1)又名“促生长因子”,在神经系统的增生、分化和神经功能的调控方面具有重要作用。研究[1-2]显示脑组织发生缺血缺氧损伤后,IGF-1及其受体表达增加,是脑卒中的血管保护因子,可促进神经元轴突生长改善神经功能[3]。另外,外源性IGF-1能够改善全脑缺血大鼠及老年小鼠的学习记忆能力,可能与其抑制海马CA1区细胞凋亡,促进海马神经细胞增生和神经元产生,提高磷脂酰肌3激酶(phosphatidylinositol3kinase,PI3K)/蛋白激酶B(proteinkinaseB,PKB)即PI3K/Akt信号通路中p-Akt、p-mTOR蛋白的表达相关[4-5]。
MAPK级联信号通路在细胞的适应、增生、分化、存活、凋亡等方面发挥重要作用。丝-苏氨酸激酶PDZ连接激酶(PDZ-binding-kinase,Pbk)是丝裂原活化蛋白激酶(mitogen-activatedproteinkinasekinase,MAPKK)家族的新成员,本课题组前期研究[6]证明在脑缺血后的神经元中Pbk的表达被抑制,而缺血后适应可以激活Pbk/p-Akt通路从而产生抗细胞氧化损伤和神经保护的作用。但是,Pbk在低氧条件下小胶质细胞和星形胶质细胞中的变化和作用尚需进一步研究。研究[7-8]显示IGF-1可能通过MAPK信号通路起到促进细胞增生,发挥神经元的去抑制作用等。同时,基于IGF-1与Akt信号通路的作用[4],提示IGF-1可能与蛋白激酶Pbk之间存在一定的关联性,但是其上下游关系尚不清楚。另外,本课题组在前期研究[9]中发现Pbk可以通过抑制组蛋白去乙酰化酶2(histonedeacetylases2,HDAC2)活性调控小胶质细胞的表型转化。这提示HDAC2与IGF-1之间也可能存在一定的关联性。因此,本实验通过培养原代星形胶质细胞和BV2小胶质细胞氧糖剥夺(oxygenandglucosedeprivation,OGD)模型探究了缺氧不同时间点后神经胶质细胞的IGF-1以及Pbk、HDAC2的表达情况,并初步探讨了三者之间的关系,为将来的机制研究提供线索。
1材料与方法
1.1实验动物与细胞
SPF级C57BL/6孕鼠,孕龄为16~18d,购于北京维通利华实验动物技术有限公司,实验动物许可证号:SCXK(京)2016-0006,小鼠小胶质BV2细胞系购于北京协和医院基础所细胞中心。
1.2主要试剂与仪器
试剂:DMEM/F12培养基购于美国Invitrogen公司,Trizol试剂、无RNA酶的水、无RNA酶的糖原购于美国Invitrogen公司,三羟甲基氨基甲烷盐酸盐[tris(hydroxymethyl)aminomethanehydrochloride,TrisHCl]、乙二胺四乙酸(ethylenediaminetetraaceticacid,EDTA)、3-(N-吗啉基)丙磺酸[3-(N-morpholino)propanesulfonicacid,MOPS]、溴化乙锭(ethidiumbromide,EB)购于华美生物工程公司,乙酸钠、甲醛、氯仿、异丙醇、100%(体积分数)乙醇购于上海化学试剂有限公司,甲醛上样染液购于美国Ambion公司,琼脂糖购于生工生物工程有限公司,RNA酶抑制剂购于美国Epicentre公司,SuperScriptTMIII反转录酶、5×RT缓冲液购于美国Invitrogen公司,2.5mmol/LdNTP混合液购于芬兰HyTest公司;2×PCR反应混合物购于美国Arraystar公司。
相关期刊推荐:怎么发医学职称论文
仪器:洁净工作台(上海博迅实业有限公司医疗设备厂)、DK-8D型电热恒温水槽(上海森信实验仪器有限公司)、GeneAmpPCR系统(德国Biosystems实验)、ViiA7RTPCR系统(德国Biosystems公司)。
1.3实验方法
1.3.1星形胶质细胞的分离
取16~18d的孕鼠分离胎鼠。放入-20℃冰箱内低温麻醉,断头前置预冷的75%(体积分数)乙醇中消毒。超净台中取出新生大鼠双侧大脑半球,置于预冷的解剖液中,在解剖显微镜下剪开颅骨及硬脑膜,迅速剥除软脑膜,并去除嗅球、基底核、海马,去除脑膜和血管组织。将收集的大脑皮质置于预冷的添加了5%(体积分数)胎牛血清的DMEM/F12中,剪碎成组织块,1000r/min离心5min,弃上清,用0.25%(质量分数)胰酶+1mmol/LEDTA的消化液于37℃消化25min,期间反复振摇2次,以血清中止消化,反复吹打使之成为单细胞悬液。离心,弃去上清,D-hank's洗涤两次,过200目的尼龙滤网,收集滤液离心后,弃上清,用完全培养液重悬细胞,接种培养瓶中,37℃、5%(体积分数)CO2培养箱中静置培养,半小时后转入新培养瓶以去除成纤维细胞。3d后首次换液去除细胞碎片。混合生长12~14d左右,细胞铺满瓶底时,旋紧瓶盖,石蜡膜封口,于37℃的恒温摇床中以250r/min的速度振摇约12~16h,收集经摇床震荡后的贴壁细胞,以D-Hank's液冲洗残留在表面的小胶质细胞和少突胶质细胞,加入新鲜的完全培养液,得到纯度较高的星形胶质细胞。
1.3.2小胶质细胞的培养
小鼠小胶质BV2细胞系购于北京协和医院基础所细胞中心,于DMEM培养液[10%(体积分数)胎牛血清+100U/mL青霉素+100μg/mL链霉素]中,在37℃、5%(体积分数)CO2培养箱中培养,观察细胞生长情况。待细胞达到70%~80%融合时,轻轻摇动培养瓶,悬浮未贴壁细胞,吸出培养液,加入PBS清洗。吸去PBS加入0.25%(质量分数)胰酶,消化1min左右,加入完全培养液终止消化。转移到15mL无菌离心管中,1000r/min离心5min。弃上清,加入适量培养液将细胞吹打成单细胞悬液,1∶3传代。
1.3.3神经胶质细胞OGD模型与分组
构建OGD模型,将细胞换至无糖培养液并置于密闭缺氧盒中,向盒内持续通入95%(体积分数)N2+5%(体积分数)CO2的混合气体5min,使盒内的空气被完全置换成不含氧气的混合气体。将缺氧盒置于37℃培养箱中,分别于OGD处理后不同时间点收集细胞。星形胶质细胞和小胶质细胞各分为4组,分别为对照组、OGD1h组、OGD3h组和OGD6h组,每组6孔。
1.3.4RT-PCR法检基因表达水平
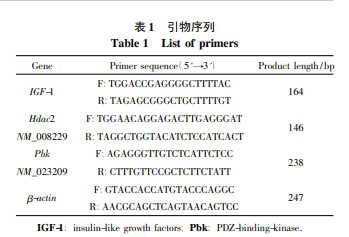
采用反转录聚合酶链反应(reversetranscriptionpolymerasechainreaction,RT-PCR)法进行检测。收集不同时间点的神经胶质细胞用Trizol法进行RNA提取,使用NanoDrop?ND-1000测定RNA浓度和纯度。根据SuperScriptTMⅢ反转录试剂盒进行cDNA合成。使用Primer5.0(英骏生物技术有限公司)设计引物,β-actinmRNA作为对照。进行实时定量PCR,引物序列详见表1。
1.4统计学方法
采用SPSS25.0以及GraphPadPrism8.0软件对数据进行处理分析及作图。计量资料以均数±标准误(x±SE)表示,多组间均数比较采用单因素方差分析(onewayANOVA),两两比较采用LSD检验;变量间相关性分析采用Pearson相关分析,以P<0.05为差异有统计学意义。
2结果
2.1OGD后小胶质细胞和星形胶质细胞IGF-1的表达情况
细胞缺氧缺糖损伤后,IGF-1在小胶质细胞中的表达水平为先升高后下降,组间比较差异有统计学意义(F=6.41,P=0.008),两两比较显示,OGD处理后6h下降较为明显,与其他组比较,差异有统计学意义(P<0.05,图1A)。IGF-1在星形胶质细胞中的表达升高,组间比较差异有统计学意义(F=3.701,P=0.029),两两比较显示,OGD处理后3h和6h明显高于对照组(P<0.05,图1B)。
2.2OGD后小胶质细胞和星形胶质细胞中Pbk的表达情况
小胶质细胞缺氧缺糖1、3和6h后Pbk的mRNA表达逐渐下降,组间比较差异有统计学意义(F=74.1,P<0.001),两两比较显示,处理后3h和6h与对照组相比,差异有统计学意义(P<0.05,图2A)。星形胶质细胞缺氧缺糖1、3和6h后Pbk的mRNA表达水平逐渐升高,组间比较,差异有统计学意义(F=14.34,P<0.001),两两比较显示,OGD处理后3h和6h与对照组相比,差异有统计学意义(P<0.05,图2B),缺氧缺糖损伤抑制了Pbk在小胶质细胞中的表达,增加了Pbk在星形胶质细胞中的表达,以OGD处理后3h和6h最为显著。
2.3OGD后小胶质细胞和星形胶质细胞中HDAC2的表达情况
小胶质细胞缺氧缺糖1、3和6h后HDAC2的mRNA表达逐渐下降,组间比较差异有统计学意义(F=27.4,P<0.001),两两比较显示,OGD处理后6h与对照组相比,差异有统计学意义(P<0.05,图3A)。星形胶质细胞缺氧缺糖1、3和6h后HDAC2的mRNA表达水平逐渐升高,组间比较差异有统计学意义(F=8.463,P=0.002),两两比较,OGD处理后3h和6h与对照组相比,差异均有统计学意义(P<0.05,图3B)。
2.4OGD后两种胶质细胞中IGF-1和信号分子Pbk、HDAC2的相关性
小胶质细胞和星形胶质细胞缺氧缺糖损伤后,信号分子Pbk和HDAC2的表达与细胞因子IGF-1的表达均呈正相关关系(P<0.05,图4)。
3讨论
本研究结果显示在缺糖缺氧的情况下,小胶质细胞中IGF-1的mRNA转录水平降低,星形胶质细胞中IGF-1的mRNA转录水平增加,且均与信号分子Pbk以及HDAC2的变化趋势相同。尽管在两种细胞中IGF-1基因表达呈相反趋势,但是在两种胶质细胞中Pbk、HDAC2均与IGF-1的转录水平呈显著正相关。
研究[10-11]显示两种胶质细胞兼具神经保护或神经毒性作用,因此分泌的细胞因子也具有不同的作用。IGF-1在缺氧缺血性脑损伤中可以参与神经形成及其髓鞘化,促进血管新生,减少细胞凋亡,起到神经保护作用[12-13]。然而其上游调节机制目前尚不明晰。
组蛋白去乙酰化酶(histonedeacetylases,HDACs)通过调控组蛋白、非组蛋白的乙酰化水平,对于基因转录的稳态维持起到重要作用。HDAC2属于经典的HADACs家族I类成员,在细胞核内与共同转录抑制子结合,从而使染色体组蛋白、非组蛋白去乙酰化而抑制转录。研究[14-15]显示HDAC2可调节细胞存活及神经元可塑性,有望作为脑卒中后神经功能恢复的治疗靶点。HDAC抑制剂(HDACinhibitor,HDACi)也被证实对脑缺血模型具有一定的神经保护作用[16-17]。在骨关节炎以及缺氧所致肺动脉高压等病理基础研究[18]中显示,组蛋白的乙酰化水平对IGF-1的表达起到调控作用。但在脑缺血缺氧性损伤中的研究尚不充分。
本研究结果提示在胶质细胞缺氧缺糖损伤早期,信号分子Pbk、HDAC2以及细胞因子IGF-1在小胶质细胞中的表达均下降,在星形胶质细胞中的表达均上升。本研究相关性分析结果提示,信号分子与细胞因子的表达呈正相关。
本课题组前期研究[6]显示Pbk在正常情况下即与HDAC1/2相结合,脑缺血再灌注损伤后随着时间的推移其结合程度增加。在小鼠脑缺血修复期,Pbk可以通过结合HDAC1/2增强其磷酸化水平,降低酶的活性,进而增加小胶质细胞中组蛋白的乙酰化水平,促进小胶质细胞从神经毒性的M1型向神经保护的M2型转化,有助于神经功能恢复,是潜在的内源性HDAC1/2抑制剂[6]。结合这一研究基础,本实验中OGD处理后小胶质细胞Pbk的表达降低,从而导致HDAC2的磷酸化水平降低,HDAC2的活性增强,抑制了组蛋白乙酰化水平,从而导致IGF-1的分泌减少;相反,OGD处理后星形胶质细胞Pbk的表达水平升高,从而导致HDAC2的磷酸化水平升高,HDAC2的活性降低,促进了组蛋白乙酰化水平,从而导致IGF-1的分泌增多。这提示在缺血缺氧早期如能尽早干预,合理提高Pbk的表达,增加Pbk/HDAC的结合程度,提高组蛋白乙酰化水平,有助于促使IGF-1的分泌,促进神经血管新生。由此笔者推测,在脑组织缺血缺氧性损伤后,可通过激活Pbk/HDAC2/IGF-1这一信号通路起到神经保护作用。
综上所述,本实验结果提示缺氧缺糖损伤显著促进了星形胶质细胞的IGF-1表达,同时抑制了小胶质细胞的IGF-1表达,Pbk及HDAC2可能参与到胶质细胞分泌神经营养因子的过程,为进一步研究缺血后神经保护机制提供了基础。未来的研究可继续探讨Pbk以及HDACs参与缺血后神经因子分泌的机制以及其他的调节通路,为临床转化提供更多可干预的分子靶点。——论文作者:王雨晴1,2陈志刚1李芳芳2张斯佳2罗玉敏2赵海苹2
